


2023.01.23 | Questions 23-24
A newborn with acidosis

Hello,
I hope you have a great week. Please feel free to email with any comments or questions. You can reach me at studyraregenetics@gmail.com.
Also, thank you to everyone who has subscribed. We are now at >200 subscribers!
Please feel free to share this post with anyone who might find it useful.
-Daniel
Questions
Question 23
A 7-day-old boy presents to the hospital with lethargy, an anion gap metabolic acidosis, and ketonuria. Biochemical testing (including serum amino acids, urine organic acids, and plasma acylcarnitine profile) is sent, revealing elevations in leucine, isoleucine, valine, and alloisoleucine in the blood. What is the most likely diagnosis in this neonate?
Question 24
The patient in question 23 is admitted to the neonatal intensive care unit for further evaluation. His newborn screen comes back positive for maple syrup urine disease, and a rapid genome sequencing test confirms the diagnosis. Which of the following is the next best step in management?
Explanation
Maple Syrup Urine Disease (MSUD, Question 23) is an inherited disorder that is characterized by the inability to break down branched-chain amino acids, including isoleucine, leucine, and valine, which leads to their accumulation in the blood. This results in a characteristic sweet “maple syrup” odor of the urine, which gave rise to the name of this disease. The presence of elevated alloisoleucine is highly specific for MSUD, while elevations in branched chain amino acids can be the result of other processes, such as a non-fasting sample. The accumulation of branched chain amino acids and corresponding alpha-ketoacids in the blood can cause an anion gap metabolic acidosis and ketonuria (see Figure below). Additionally, the accumulation of these amino acids and alpha-ketoacids in the brain can cause neurological symptoms, such as seizures or lethargy, the latter of which is seen in this patient. Note that ketonuria in a newborn, as seen in this patient, should always prompt concern for an underlying metabolic disorder.
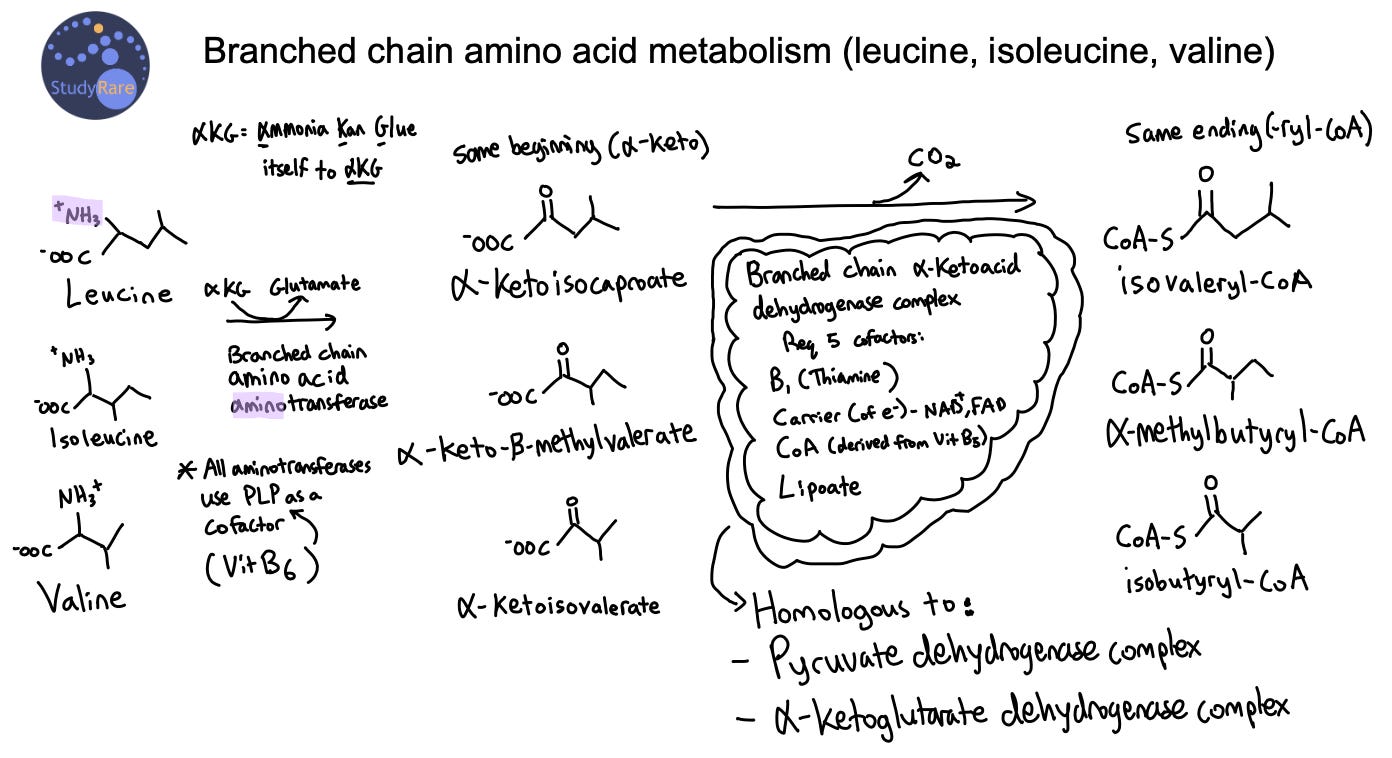
MSUD can be categorized as classic (most severe), intermediate, or intermittent (most benign). The initial presenting symptoms vary, though many patients are initially detected on newborn screening. Classic MSUD presents in the neonatal period with signs of metabolic intoxication (irritability, hypersomnolence, & difficulty feeding) on day of life (DOL) 2-3 after an initial asymptomatic period. From DOL 4-6, patients present with progressive lethargy, apnea, back arching, and "bicycling" movements. Shortly after the first week of life, patients develop cerebral edema, coma, and respiratory failure, which can lead to death if untreated. However, with early and sustained treatment, patients with classic MSUD achieve most major developmental milestones, but may have some developmental delay and reduced cognitive function relative to unaffected peers. Mood disorders, ADHD, and movement disorders (mainly tremor or dystonia) in adulthood are also common among patients with MSUD. Think “Movement disorder, mood Swings, & Developmental delay in MSUD.”
Patients with classic MSUD are also at risk for acute metabolic decompensation in the setting of illness, fasting, or stress, with the risk being highest in the first few years of life. Infants and toddlers present with altered mental status, acute dystonia, and ataxia, while older individuals present with psychiatric disturbances (e.g. hallucinations), neurologic deficits (e.g. ataxia, choreoathetosis), and cognitive impairment. One particularly worrisome complication is cerebral edema, though the mechanism behind this is not completely understood.
💡 Think: “patients with MSUD can have SUDden decompensation” and “the brain Might Swell in MSUD.”
During fasting or stress, proteins are broken down into amino acids (including branched chain AA), which are then converted into Krebs cycle intermediates that make cellular energy. However, in MSUD, alpha-ketoacids (see Figure above, center column) accumulate when the body attempts to catabolize BCAAs, leading to a metabolic crisis. Intoxication is primarily driven by the amino acid leucine and its corresponding alpha-ketoacid, alpha-ketoisocaproic acid. This is due in part from leucine’s ability to interfere with transport of other large neutral amino acids across the blood-brain barrier.
Incorrect answers
Question 23: Classic phenylketonuria, methylmalonic acidemia (MMA), and propionic acidemia (PA) are not characterized by elevations in BCAAs or alloisoleucine. MMA and PA can present with acidosis in neonates after an initial asymptomatic period, therefore sharing some of the clinical aspects of MSUD.
Question 24: A leucine-restricted diet is the mainstay of treatment in MSUD. BCAA-free medical foods, dietary protein restriction, and supplementation with isoleucine and valine are also indicated. Thiamine supplementation (not niacin) can be attempted and is effective in some cases of MSUD. This is because the BCKD complex requires thiamine as a cofactor. Tyrosine supplementation is not indicated in MSUD.
Learning objective
MSUD is a disorder of branched chain amino acid metabolism that is due to mutations in the branched chain alpha-ketoacid dehydrogenase complex. Classic MSUD presents in the first week of life with lethargy and metabolic acidosis. Patients are at risk of metabolic decompensation in the setting of stress or fasting. Treatment is with dietary leucine restriction, protein restriction, BCAA-free medical foods and formula, and thiamine.
2023 ABMGG General Exam Blueprint | V. Single Gene Inheritance → d. Single Gene Disorders → ix. Metabolic disease → 1. Amino acid disorders (page 3)