


2023.03.05 | Questions 34-35
Cancer Genetics (2/3) - A family with neurofibromatosis type 1

Hello,
This is the 2nd in a series of 3 posts related to cancer genetics. This post focuses on NF1, the most common genetic cancer predisposition syndrome.
Please feel free to email with any comments or questions. My email is studyraregenetics@gmail.com.
Also, please share this post with any colleagues who might find it useful!
-Daniel
Questions
Question 34
A 10-year-old boy with neurofibromatosis type 1 (NF1) presents with a painless mass on his right neck. The mass has been slowly growing over the past 3 years. The physician notes a ropy texture on palpation of the mass. MRI of the cervical spine shows a large tumor that is deemed inoperable. Given his diagnosis of NF1, the tumor most likely originated from which tissue type?

Question 35
The patient in Question 34 achieves tumor shrinkage with the appropriate therapy. He presents several months later for a follow-up visit with his 36-year-old mother, who also has a clinical diagnosis of NF1. She has not received regular follow-up and her last visit with her PCP was 15 years ago. Based on her diagnosis of NF1, she should begin cancer screening with which of the following modalities?
Explanations
Answer to Q34: Peripheral nerve
Answer to Q35: Mammography
Neurofibromatosis type 1 (NF1) is a neurocutaneous syndrome, with ‘neuro’ referring to brain/nerves and ‘cutaneous’ referring to skin. The most common features of NF1 include café au lait macules (CALMs), freckling in the axillae or groin, neurofibromas (which can be cutaneous, subcutaneous, or plexiform), and Lisch nodules (small, benign, pigmented iris tumors). There is also an increased risk of several malignancies (discussed below), hypertension, learning disabilities, and seizures. Bony lesions such as sphenoid wing dysplasia or tibial pseudoarthosis may also be seen. NF1 is a clinical diagnosis with multi-systemic diagnostic criteria that are described in the chart below. In patients with a clinical diagnosis of NF1, genetic testing can confirm the diagnosis and helps distinguish between NF1 and Legius, a related syndrome (discussed below).

NF1 is the most common cancer predisposition syndrome
NF1 affects 1 in 3000 individuals and predisposes to benign tumors as well as malignant cancers. Malignant cancers associated with NF1 include malignant peripheral nerve sheath tumors (MPNST), leukemia, pheochromocytoma, and breast cancer. Adult females with NF1 under age 50 are at an increased risk of breast cancer. The absolute risk of breast cancer in females with NF1 is 20%–40%, compared to ~12% in the general population. Because of this risk, the National Comprehensive Cancer Network recommends that women with NF1 start annual breast cancer screening with mammography at age 30 rather than at age 40 for women at average risk (Question 35).
Patients with NF1 also are predisposed to develop benign tumors known as neurofibromas, which arise from the Schwann cells that myelinate peripheral nerves (Question 34). There are 3 types of neurofibromas: cutaneous (most common, found on skin), subcutaneous, and plexiform. The patient in Question 34 has an inoperable plexiform neurofibroma (PN) on his neck, which can present with a ‘ropy’ or ‘bag of worms’ texture due to involvement of multiple nerves. A minority of plexiform neurofibromas transform into MPNST, which are invasive tumors that may present as a rapidly-enlarging and painful mass. Selumetinib is a recently-approved therapy indicated for non-resectable plexiform neurofibromas and would be an appropriate therapy for the patient in Question 34 given his clinical presentation.
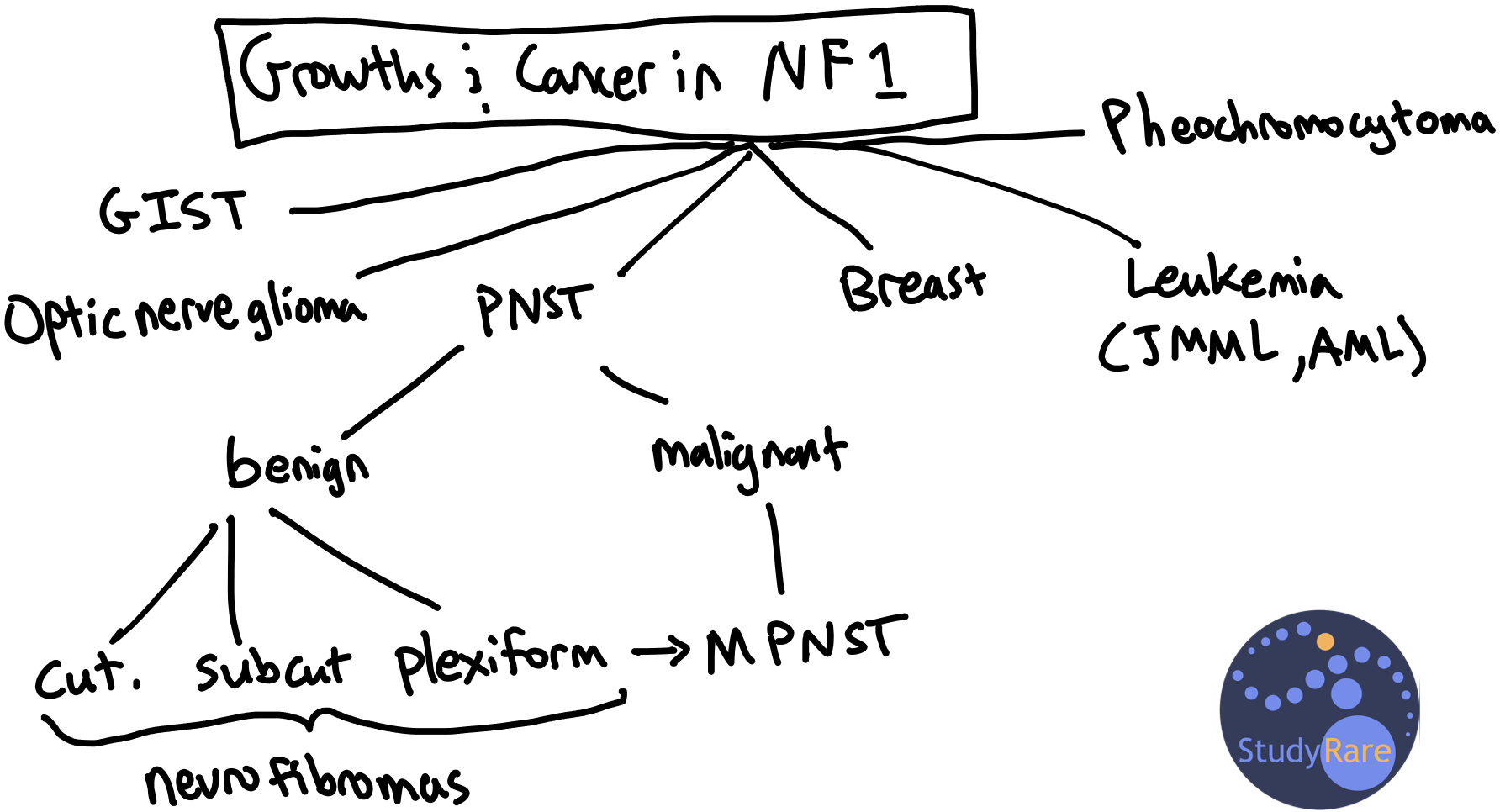
💡 To remember the types of growths and cancers associated with neurofibromatosis 1, think “PhIBBBrOMa:” Pheochromocytoma, Iris tumors (i.e. Lisch nodules), Blood (leukemia), Bowel (i.e. GI stromal tumor), Breast, Optic nerve glioma, Malignant PNST.
Molecular genetics & inheritance of NF1
Neurofibromatosis type 1 (NF1) is caused by heterozygous mutations in the NF1 gene, which is located on chromosome 17q11.2. Most pathogenic variants in NF1 are truncating (eg frameshift, nonsense, and splice-site mutation), though multi-gene deletions involving NF1 and causing a more severe phenotype have been reported. The NF1 gene encodes for the protein neurofibromin, which is a GTPase-activating protein that negatively regulates the RAS/MAPK signaling pathway. Thus, when neurofibromin is not functioning properly or present in sufficient quantity (due to an NF1 mutation), there is increased signaling through the RAS/MAPK pathway, which in turn promotes cellular growth. NF1 is classified as a RASopathy, a family of disorders that are caused by mutations that alter signaling through the RAS/MAPK pathway and that includes Noonan syndrome and cardio-facio-cutaneous syndrome.
NF1 is inherited in an autosomal dominant manner. Approximately 50% of NF1 cases are de novo mutations, while the other 50% of cases are inherited from an affected parent.
💡 NF1 is New (de novo) in Fifty percent of cases.
Complications with diagnosing NF1
There are two key elements that complicate both the molecular and clinical diagnosis of NF1. First, the NF1 gene has multiple pseudogenes, which are a non-functional copies of the gene that can lead to false positive or false negative sequencing results. If possible, samples should be sent to a lab with expertise in sequencing NF1, such as the molecular lab at UAB. Sequencing of NF1 is typically performed at a high read depth, so as to detect any mosaic variants in NF1. Second, there is a clinical syndrome called Legius syndrome (due to mutations in SPRED1) that overlaps clinically with NF1. Patients with Legius syndrome have skin pigmentary findings that meet the diagnostic criteria of NF1 (i.e., ≥6 CALMs and axillary or inguinal freckling), but lack most other features (e.g. neurofibromas, optic pathway gliomas) seen with NF1. Therefore, when the diagnosis of NF1 is made based on cutaneous findings, our usual practice is to send concurrent molecular testing for both NF1 and SPRED1.
There have been some genotype-phenotype correlations, such as a multi-gene deletion including NF1 being associated with larger numbers and earlier appearance of cutaneous and plexiform neurofibromas, a greater risk of MPNST, and more severe cognitive abnormalities. However, NF1 exhibits a high degree of variable expressivity, even within families with the same genetic variant, and it is therefore difficult to predict the overall severity of disease or the age of symptom onset given a patient’s mutation. This may change in the future as we learn more about the genetic basis of NF1 and the contribution of genetic modifiers.
Learning objective
Neurofibromatosis type 1 (NF1) is a disorder caused by variants in the NF1 gene and characterized by multiple CALMs, cutaneous neurofibromas, and learning disability. Neurofibromas, a hallmark feature of NF1, arise from the axon sheaths (Schwann cells) of peripheral neurons. Patients with NF1 are predisposed to a variety of malignancies including breast cancer, MPNST, leukemia, and pheochromocytoma. Earlier breast cancer screening beginning at age 30 is recommended for adult women with NF1.
2023 ABMGG General Exam Blueprint | V. Single Gene Inheritance → d. Single Gene Disorders → vi. Neurocutaneous disorders (page 3)