

2023.03.21 | Questions 39-40
2023.03.21 | Questions 39-40
Metabolic masquerading (1/3) - A boy with cerebral palsy

Hello,
Congratulations to all med students who matched last week! 🎉 If you know of anyone who matched into a genetics program, feel free to forward this email to give them a head start!
This is the 1st in a series of 3 posts titled “metabolic masquerading.” There are a number of metabolic disorders that may be misdiagnosed as “cerebral palsy,” “recurrent abdominal pain,” “clumsiness,” etc. Some of these disorders, such as the one discussed today, have treatment options that can improve symptoms, so they are important to recognize and diagnose.
Please feel free to email with any comments or questions about this post (daniel@studyrare.com). Have a great week!
-Daniel
Questions
Question 39
A 8-year-old boy with cerebral palsy presents with progressive tightness of the lower limbs over the past several years. He was born at term and has no history of birth trauma. He started walking at 20 months of age, and his other developmental milestones have been normal. His lower limb stiffness is worse in the evenings and improves in the morning after sleeping. Physical exam reveals bilateral lower limb spasticity, brisk lower extremity reflexes, and a scissoring gait. A brain MRI is normal. He is trialed on low-dose levodopa/carbidopa with substantial improvement in spasticity and gait. He most likely has which of the following disorders?
Question 40
Genetic testing is performed and confirms the diagnosis of GTP cyclohydrolase I deficiency. Which of the following types of genetic variants is most likely to be found in the GCH1 gene?
Explanations
A child with long-standing lower-extremity spasticity and a scissoring gait (signs of dystonia) suggests a clinical diagnosis of a type of cerebral palsy called spastic diplegia. While the underlying cause of cerebral palsy remains unknown in many cases, the progressive nature of his symptoms are not characteristic of cerebral palsy, which is typically static and non-progressive. The fact that the patient’s spasticity responded well to levodopa/carbidopa, along with the diurnal 🌓☀️ fluctuation in symptoms with improvement after sleep and normal cognition, is more characteristic of a dopa-responsive dystonia (DRD). In this patient, DRD was masquerading as ‘cerebral palsy.’ Brain MRI is also typically normal in patients with DRD (as is the case with this patient) in contrast to other disorders (e.g. leukodystrophy) that may present with progressive dystonia.
DRD classically presents in childhood or adolescence with progressive dystonia, mild parkinsonism, marked diurnal fluctuations, and improvement with sleep. However, there is a broad clinical spectrum, with some patients not presenting until mid- or late adulthood. Therefore, a diagnosis of DRD should be considered in both pediatric and adult patients with lower-extremity spasticity (particularly if progressive and not present at birth) who do not fit the typical clinical picture of cerebral palsy. Biochemical testing for suspected DRD (see table below) includes checking urine or blood pterins (e.g. neopterin, biopterin) and DHPR activity.
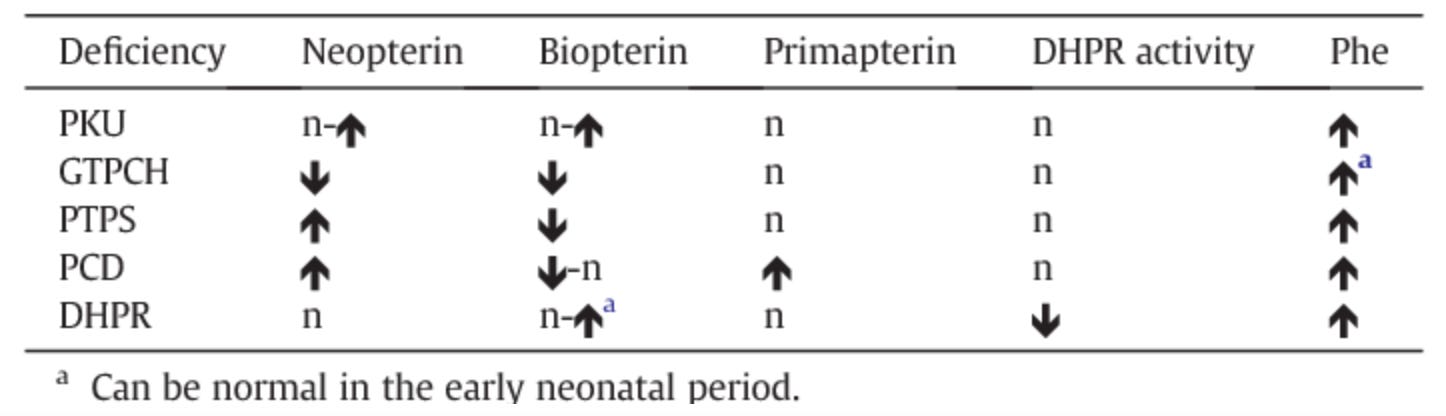
Biochemistry of DRD
DRD is most commonly caused by a deficiency in the enzyme GTP cyclohydrolase I (GTPCH1, encoded by the gene GCH1) (Question 39). GTPCH1 catalyzes the first of three enzymatic steps in synthesizing tetrahydrobiopterin (BH4), an important cofactor for several hydroxylases, including tyrosine hydroxylase (TH). TH converts tyrosine into L-DOPA, a precursor to dopamine (see figure below). Therefore, a deficiency in the enzymes within the BH4 synthesis or recycling pathway reduces the amount of dopamine in the brain. The lack of dopamine causes a movement disorder (dystonia) that responds clinically to levodopa (i.e. L-DOPA, a precursor to dopamine) and carbidopa (prevents peripheral breakdown of levodopa), hence the term ‘dopa-responsive’ dystonia). Dopamine depletion or loss of dopaminergic neurons in certain regions of the brain are an established cause of dystonias and can be seen in other movement disorders, most notably Parkinson’s disease, that may also respond to levodopa/carbidopa.
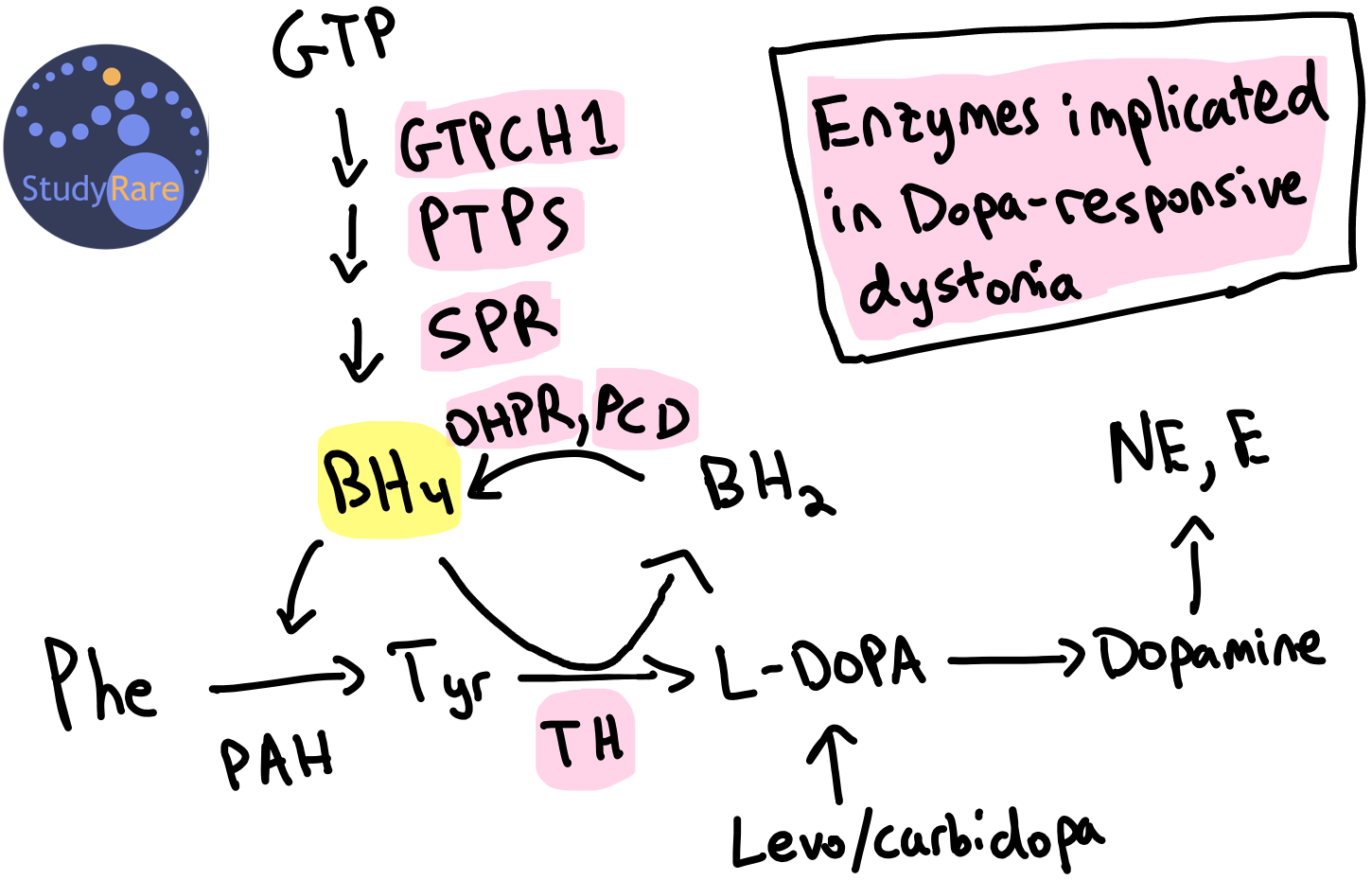
Genetic variants and inheritance of GTPCH1 deficiency
The most common causes of GTPCH1-related DRD are heterozygous missense or nonsense (e.g. stop-gain) variants in the GCH1 gene that result in haploinsufficiency (Question 40). As explained above, this leads to reduced levels of BH4 and a subsequent reduction in dopamine production. These variants in GCH1 can be de novo or inherited in an autosomal dominant manner. In rare cases, GTPCH1 deficiency can be also inherited as an autosomal recessive disorder that is associated with severe symptoms in infants including global developmental delay, seizures, and regression that does not respond to levodopa/carbidopa.
Incorrect answers (Q39)
Arginase deficiency is a urea cycle disorder that is characterized by episodic hyperammonemia. While progressive spasticity can be a feature of undiagnosed arginase deficiency, it is usually associated with other features such as global developmental delay, intellectual disability, seizures, and episodic hyperammonemia. Furthermore, improvement of spasticity with levodopa/carbidopa is not characteristic of arginase deficiency. Treatment of arginase deficiency is with restriction of dietary protein and oral nitrogen-scavenging medications.
Biotinidase deficiency can present in a school-age child or adolescent with motor limb weakness, spastic paresis, and decreased visual acuity. Holocarboxylase synthetase deficiency, a related biotin processing disorder, can also present with progressive spasticity. Both disorders are typically detected in the U.S. through newborn screening. Symptoms respond to biotin supplementation, not levodopa/carbidopa.
Spastic paraplegia 4 (SPAST-HSP) is due to autosomal dominant mutations in SPAST and can present with progressive spasticity in the lower extremities. While symptoms can begin at any age, the diagnosis is most often made in young adults. Improvement with levodopa/carbidopa is not a feature of SPAST-HSP. Treatment is supportive.
Incorrect answers (Q40)
Imprinting defects, which refer to the loss of normal epigenetic marks on a gene, are not known to cause GCH1 deficiency. Beckwith-Wiedemann syndrome and Angelman syndrome are examples of syndromes that are caused by imprinting defects. Trinucleotide repeat expansions are also not a known mechanism of disease in GCH1 deficiency. However, trinucleotide repeat expansions are associated with a variety of movement disorders including spinocerebellar ataxia, Friedreich’s ataxia, and Huntington's disease.
Learning objective
Dopa-responsive dystonia (DRD) is most commonly caused by GTP cyclohydrolase I deficiency and presents with lower-extremity spasticity and dystonia that improves with sleep and with low doses of levodopa/carbidopa. There are several disorders, including DRD, arginase deficiency, and biotinidase deficiency, that can be (mis)diagnosed as cerebral palsy and have treatment options. A patient with progressive spasticity with onset after the neonatal period always deserves further evaluation — don’t settle for a diagnosis of “cerebral palsy.” You could have a treatable disorder on your hands!
2023 ABMGG General Exam Blueprint | V. Single Gene Inheritance → d. Single Gene Disorders → ix. Metabolic disease (page 3)
2023 ABMGG Biochemical Genetics Exam Blueprint | XII. Neurotransmitters → b. Disorders of dopamine synthesis (page 4)
References (and some other examples of metabolic masquerading)
Holocarboxylase synthetase deficiency: a treatable metabolic disorder masquerading as cerebral palsy
Biotinidase deficiency: A treatable cause of hereditary spastic paraparesis
Neurogenetic and Metabolic Mimics of Common Neonatal Neurological Disorders
Fifteen-minute consultation: Red flags for metabolic disease in routine bloods