

2023.04.06 | Questions 43-44
2023.04.06 | Questions 43-44
Metabolic masquerading (3/3) - A newborn with HIE

Hello,
This is the 3rd in a series of 3 posts titled “metabolic masquerading.” There are a number of metabolic disorders that may be misdiagnosed as “cerebral palsy,” “recurrent abdominal pain,” “clumsiness,” etc. Some of these disorders, such as the ones in this series, have treatment options that can improve symptoms, so they are important to recognize and diagnose.
Please feel free to share this post with anyone who might find it useful, and reach out with any feedback or comments (daniel@studyrare.com).
-Daniel
Questions
Question 43
A 1-day-old boy born at term is noted to have jitteriness and intermittent stiffening of the limbs. APGAR scores were 8 and 9, and there was no birth trauma. A brain MRI shows global injury of the white matter, basal ganglia, and thalamus, raising concern for hypoxic–ischemic encephalopathy (HIE). An EEG shows a burst suppression pattern, and he is started on anti-epileptic medications. Laboratory evaluation shows decreased uric acid, increased hypoxanthine and xanthine, and elevated urine S-sulfocysteine. What is the most likely diagnosis?
Question 44
Whole genome sequencing is sent and shows biallelic, pathogenic variants in MOCS1, confirming the diagnosis of molybdenum cofactor deficiency. Which treatment modality has been shown to extend the life of patients with this disorder?
Explanation
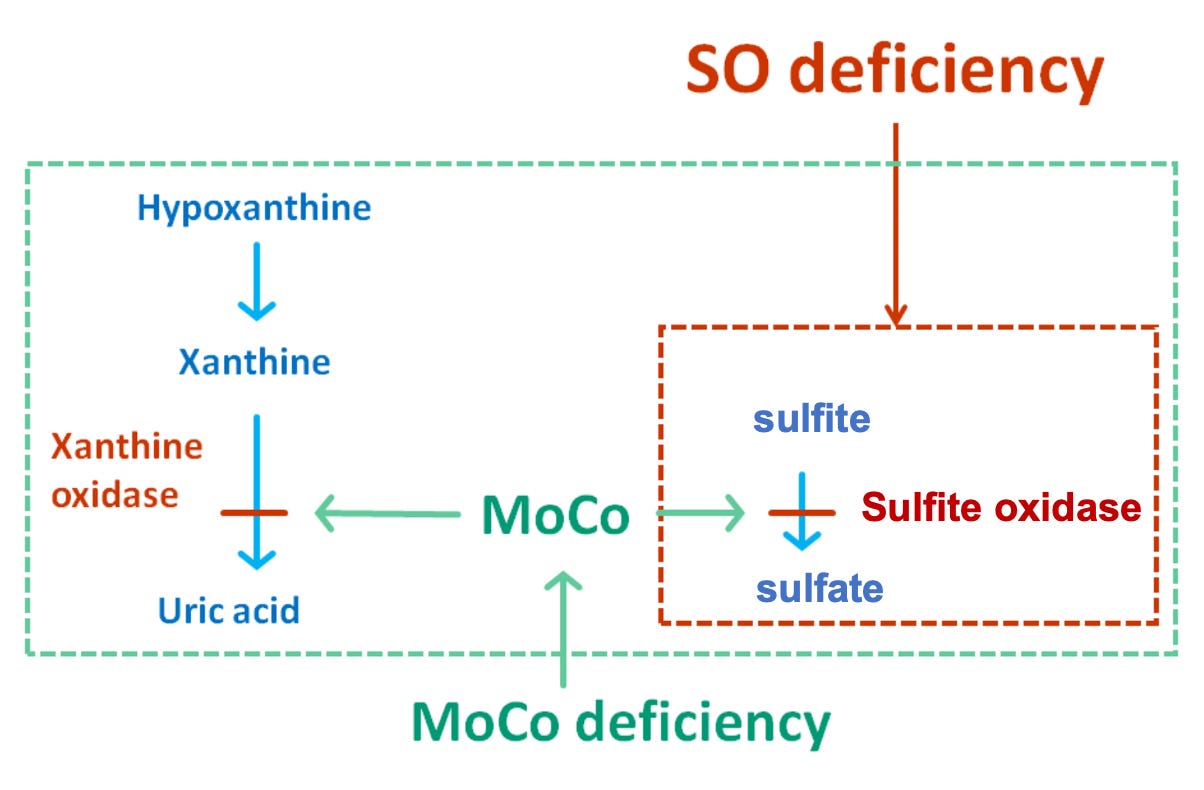
The clinical presentation and laboratory findings in this patient suggest a diagnosis of molybdenum cofactor deficiency (MoCD) (Question 43). MoCD is an autosomal recessive disorder caused by a defect in the molybdenum cofactor (MoCo) biosynthesis pathway (explained further below). Patients with MoCD typically present in the neonatal period with seizures, poor feeding, and developmental delay. The brain MRI findings in patients with MoCD may resemble the MRI findings in hypoxic ischemic encephalopathy (HIE), a condition affecting ~1 in 1000 term infants that results from impaired oxygen delivery to the brain that causes decreased alertness, abnormal movements, seizures, and abnormal muscle tone. Laboratory findings in MoCD reflect deficiencies in two MoCo-dependent enzymes. First, deficiency of sulfite oxidase leads to accumulation of toxic levels of sulfite, which is excreted as S-sulfocysteine in the urine. Second, deficiency of xanthine oxidase leads to accumulation of xanthine and hypoxanthine, as well as low uric acid levels. These laboratory values are important to know for board examinations and also in clinical practice.
What is molybdenum cofactor (MoCo)?
Molybdenum is a metal, just like iron, copper, or zinc. Enzymes use metals to help transfer electrons between chemical compounds, among other chemical reactions. Molybdenum is complexed together with a pterin to form the molybdenum cofactor (kind of like how iron and protoporphyrin are complexed together to form heme). Pterins are chemical moieties that are found in certain cofactors including riboflavin (vitamin B2), folate (vitamin B9), and tetrahydrobiopterin (BH4).
MoCo synthesis
There are 4 genes involved in converting GTP into MoCo. Pathogenic variants in any of the 4 genes that convert GTP into MoCo result in MoCD. The most commonly affected gene in this pathway is MOCS1, which encodes the enzyme that catalyzes the first step in the pathway and converts GTP to cyclic pyranopterin monophosphate (cPMP).
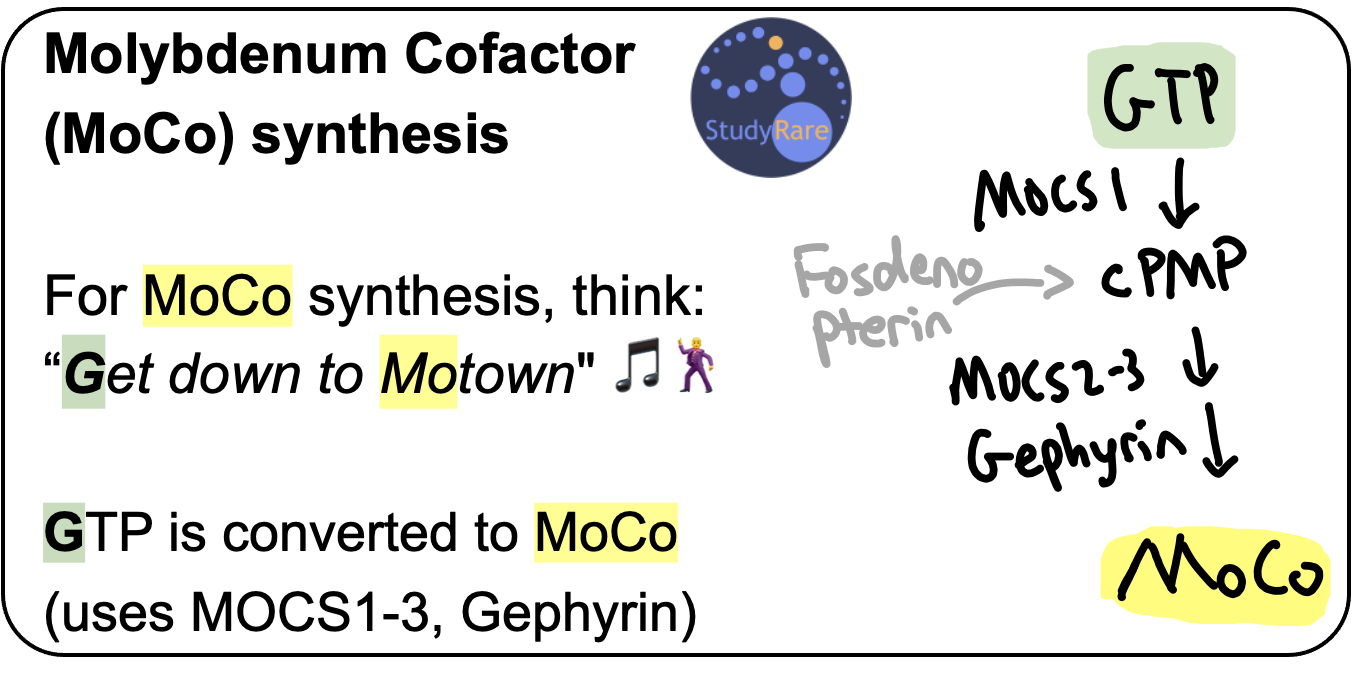
Treatment of MoCD due to MOCS1 deficiency
Fosdenopterin, a synthetic analog of cPMP, is FDA-approved to treat patients with pathogenic variants in MOCS1 by replacing the depleted product, cPMP (Question 44). This treatment improves survival in patients with MoCD due to MOCS1 deficiency, particularly when initiated early. The survival benefit here is noteworthy, as many drugs for rare diseases are now being approved based on surrogate endpoints (eg biomarkers) or other endpoints that are less clinically meaningful (eg timed walk tests).
MoCo is a cofactor for 3 enzymes. Remember them as “SAX” 🎷.

Sulfite oxidase (SOX) is a mitochondrial enzyme that catalyzes the conversion of sulfite (toxic) to sulfate (less toxic, more readily excreted). This reaction removes the sulfite produced during the metabolism of sulfur-containing amino acids. Deficiency of SOX leads to the accumulation of toxic levels of sulfite in various tissues, including the brain, which causes severe neurological damage. Patients who present with isolated sulfite oxidase deficiency may also present similarly to MoCD. One of the characteristic biomarkers of this disorder, S-sulfocysteine, is produced by reaction of sulfite with the amino acid cystine and can be detected in the urine. Remember the phrase “MUSULE” – In MoCo, Urine S-SULfocysteine is Elevated.
Aldehyde oxidase (AOX) is important for the metabolism of certain drugs. Deficiency of AOX does not contribute to the accumulation or deficiency of a detectable biomarker.
Xanthine oxidase (XOX) converts hypoxanthine to xanthine, and also xanthine to uric acid. This reaction is important for the metabolism of purine nucleotides. Deficiency of XOX leads to increased levels of hypoxanthine and xanthine, as well as decreased levels of uric acid. Accumulation of xanthine can also lead to the formation of xanthine stones in the kidney.
Incorrect answers (Question 43)
Classic non-ketotic hyperglycinemia (NKH) results in the accumulation of glycine in the central nervous system, leading to treatment-refractory seizures and major developmental delays. Patients have elevated glycine levels in the CSF. On occasion, NKH can masquerade as HIE. However, the laboratory findings described in the question stem (decreased uric acid, increased hypoxanthine and xanthine, and elevated urine S-sulfocysteine) are not consistent with the diagnosis of NKH.
Zellweger spectrum disorders (ZSD) affect the metabolism of lipids and/or the formation of peroxisomes, leading to severe neurological symptoms that could mimic HIE. Patients with ZSD have elevated very long chain fatty acids, pristanic acid, and pipecolic acid, along with deficient plasmalogens in erythrocytes. The laboratory findings described in Question 43 are not consistent with this diagnosis.
Pyruvate carboxylase deficiency (PCD) can present in the neonatal period with symptoms that include rigidity, tremor, and seizures and may resemble HIE. Patients may have an elevated lactate/pyruvate ratio, elevated citrulline, elevated ammonia, and low glutamine. The laboratory findings described in the question stem are not consistent with this diagnosis.
Incorrect answers (Question 44)
The incorrect answer choices provided in this question can be used to manage other metabolic disorders. While liver transplantation (LT) is not a recognized treatment for molybdenum cofactor deficiency, LT can be used to treat other metabolic disorders, such as those involving the urea cycle and certain glycogen storage diseases. Sodium phenylbutyrate is approved for the treatment of urea cycle disorders, but it does not address the underlying metabolic defect in molybdenum cofactor deficiency. Thiamine (Vitamin B1) is not a recognized treatment for MoCD but can be used to treat certain disorders, such as thiamine-responsive megaloblastic anemia (SLC19A2).
References
Inborn errors of metabolism masquerading as hypoxic-ischemic encephalopathy
Learning objective
Molybdenum cofactor deficiency (MoCD) is caused by a defect in the molybdenum cofactor (MoCo) biosynthesis pathway, leading to a deficiency of several enzymes, including sulfite oxidase and xanthine oxidase. Patients with MoCD typically present in the neonatal period with seizures, poor feeding, and developmental delay that can mimic HIE. Fosdenopterin, a synthetic analog of cyclic pyranopterin monophosphate (cPMP), has been shown to extend the life of patients with MoCD due to MOCS1 deficiency.
2023 ABMGG General Exam Blueprint | V. Single Gene Inheritance → d. Single Gene Disorders → ix. Metabolic disease (page 3)