

2023.09.04 | Questions 58-59
2023.09.04 | Questions 58-59
Neurogenetics (1/3) - A boy with intellectual disability and fractures

Hello,
Happy Labor Day! This week’s questions highlight an intellectual disability syndrome with a propensity for fractures and a unique biochemical signature. I also uploaded a video related to the topic of this newsletter post. This is the first of 3 posts in a series on neurogenetics.
Please feel free to reach out with any comments, questions or ideas (daniel@studyrare.com). Also, if you recently took the ABMGG or ABGC board exam and are open to sharing your experience preparing for these exams, I still have some availability for 15-minute 1:1 meetings this week. Feel free to sign up here.
If you find this content valuable and would like to support my work, consider buying me a coffee.
Have a great week!
-Daniel
Questions
Question 58
A 7-year-old boy with intellectual disability presents to the neurogenetics clinic for an evaluation. He has a history of several fractures after minor falls. Family history is notable for a maternal uncle with intellectual disability. Physical exam reveals asymmetric facies with a prominent lower lip and a high arched palate, a thin appearance, and moderate hypotonia. A DEXA scan shows osteoporosis. Metabolomic profiling of the patient shows elevations in ornithine and spermidine. Which of the following is the most likely diagnosis?
Question 59
Genetic testing is sent for the individual in Question 58 and reveals a pathogenic loss-of-function variant in the gene spermine synthase (SMS). What is the most likely inheritance pattern of this disorder?
Explanations
Q58: Snyder-Robinson syndrome
Q59: X-linked recessive
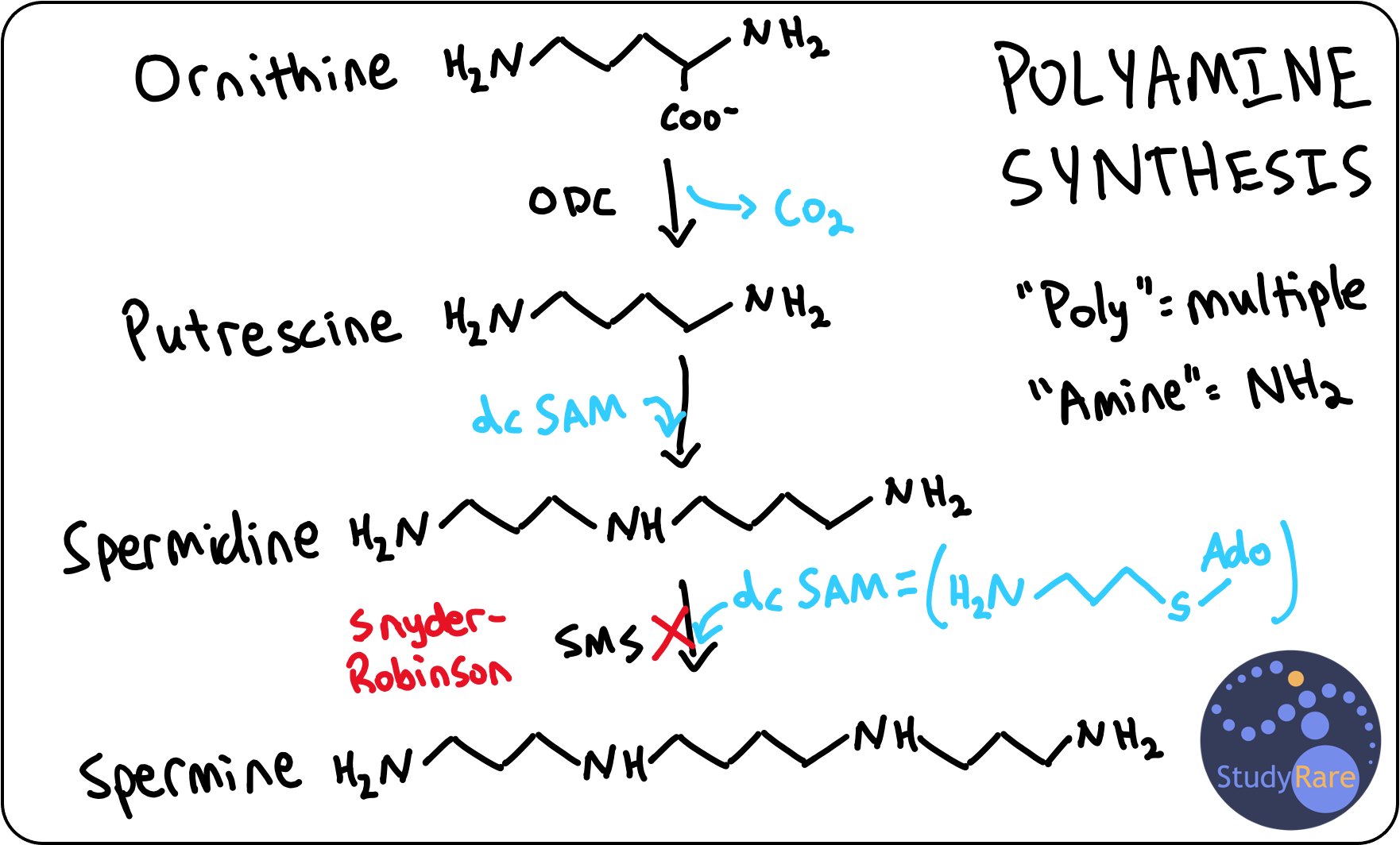
Snyder-Robinson syndrome (SRS), also known as spermine synthase deficiency, is an X-linked recessive intellectual disability syndrome (suggested by the presence of an affected maternal uncle in Q58) that is characterized by osteoporosis, seizures, and facial dysmorphism (e.g. asymmetric facies, prominent lower lip, and high-arched palate). Patients also often have a thin, frail build with low muscle mass and hypotonia, as was described in this patient. Patients with SRS typically develop osteoporosis within the first decade of life (think “Ro-bone-son”🦴), which can result in fractures with minimal or no trauma. Management is primarily supportive and includes physical and occupational therapy for hypotonia, calcium supplementation (to help improve bone density), and anti-epileptics to control seizures.
SRS is definitively diagnosed with molecular testing, which shows a hemizygous loss-of-function variant in spermine synthase (SMS). In this patient, metabolomic profiling shows elevated ornithine and spermidine levels, which are highly characteristic of SRS. Of note, spermidine levels are not typically ordered as part of routine biochemical analyses (even by biochemical geneticists), though metabolomic profiling (like an “exome for metabolites”) is one way that elevated spermidine could be detected.
SRS is characterized as a polyamine synthesis disorder. Polyamines are compounds composed of many (“poly-”) amine (“-NH2” or “-NH-”) groups that support a variety of cellular processes, including cell division, cell proliferation, and DNA condensation in sperm. The three main polyamines in humans are putrescine, spermidine, and spermine. Spermine synthase is the last enzyme in the polyamine biosynthesis pathway (see diagram below for details) and converts spermidine to spermine. This diagram also makes it easier to understand why patients with SRS have elevated spermidine levels.
💡Spermine is so named because it was initially discovered in sperm cells. This can help you remember that spermine synthase deficiency is X-linked recessive and thus primarily affects males (because males produce sperm).
Incorrect answers (Q58)
Ornithine aminotransferase (OAT) deficiency is an autosomal recessive disorder that manifests with myopia and night blindness 👀. While OAT deficiency can present with elevated ornithine levels, it is more commonly associated with gyrate atrophy of the choroid and retina and does not typically result in osteoporosis or intellectual disability. 💡Remember, patients with OAT need an OphthAlmologisT.
Ornithine decarboxylase (ODC) superactivity syndrome (aka Bachmann-Bupp syndrome) is an autosomal dominant disorder due to gain-of-function mutations in the ornithine decarboxylase enzyme (step 1 in the polyamine synthesis pathway). ODC superactivity leads to excess putrescine and is not associated with elevated ornithine as seen in this case. Patients with ODC superactivity present with global developmental delay, alopecia, macrocephaly, facial dysmorphism, and variable white matter abnormalities on neuroimaging.
Ornithine translocase deficiency, also known as Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is an autosomal recessive urea cycle disorder disorder caused by mutations in SLC25A15, which encodes for the ornithine translocase. While HHH syndrome presents with elevated ornithine, patients would not have elevated spermidine levels. In addition, intellectual disability may be a feature of HHH though osteoporosis and facial dysmorphism are not characteristic of HHH.
💡Do not confuse ornithine translocase deficiency with ornithine transcarbamylase deficiency, an X-linked urea cycle disorder that manifests with hyperammonemia and low citrulline levels.
Learning objective
Snyder-Robinson syndrome (SRS) is an X-linked recessive intellectual disability syndrome characterized by osteoporosis, facial dysmorphism, and seizures. SRS is due to a biochemical defect in the polyamine synthesis pathway, which is important for a variety of biological processes including cell proliferation, cell division, and DNA condensation. Laboratory findings for SRS include elevated ornithine and elevated spermidine. Management of SRS is supportive.
2023 ABMGG General Exam Blueprint | V. Single Gene Inheritance → d. Single Gene Disorders → ix. Metabolic disease
2023 ABGC Exam Content Outline | Domain 1. Clinical Information, Human Development, and Genetic Conditions → C. Genetic Conditions → 1. Clinical features & 7. Mode of inheritance