


2023.10.17 | Questions 67-68
Ophthalmic genetics 👁️ (2/3) - An adult with ptosis (& Feb 2024 ABGC board review course!)

Hello,
This is the 2nd in a series of 3 posts related to ophthalmic genetics. This week’s questions discuss a case of a middle-aged man with ptosis and difficulty swallowing. I also uploaded a YouTube video related to the topic of this post.
A few additional points: I will be at the NSGC meeting in Chicago this upcoming week Oct 17 - 21. I am planning to attend the student and first-time attendee events starting with the welcome orientation on Tuesday evening. If you are attending any of these events, please say hello. I would love to meet you!
Also, if you are planning to take the ABGC exam in February 2024, I am hosting a 25-hour board review bootcamp that is tailored to the content on the new ABGC exam. The course will take place from November 2nd, 2023 - January 12th, 2024. You can sign up here (also see flyer below).

If you would like to support my work, please share this newsletter with genetic counseling students, clinical genetics fellows, or anyone interested in learning more about genetics and rare disease. You can also consider buying me a coffee.
Please feel free to reach out with any feedback or ideas for future posts (daniel@studyrare.com).
Have a great week!
-Daniel
Questions
Question 67
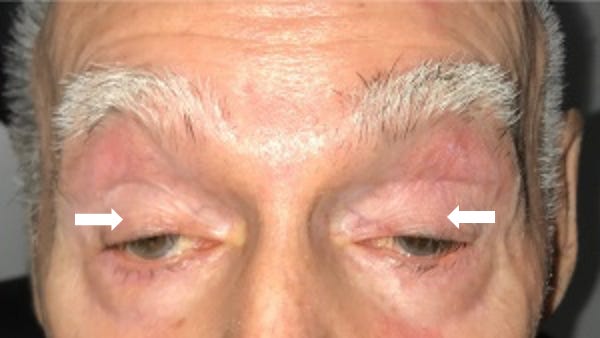
A 56-year-old man presents with droopy eyelids and trouble swallowing food, which has been getting stuck in his throat. Both symptoms have been getting worse over the past year. He also reports unintentional weight loss of 15 pounds over the past 6 months and was recently hospitalized with aspiration pneumonia. Physical exam reveals bilateral ptosis. His vision is 20/20 and he has no visual field defects. His mother also had similar symptoms that also started in her mid-50’s. A whole exome sequencing study including mitochondrial DNA sequencing is non-diagnostic. What is the most likely diagnosis?
Question 68
The disorder affecting the patient in Question 67 is inherited in which of the following manners?
Explanation
Q67: Oculopharyngeal muscular dystrophy
Q68: Autosomal dominant
Oculopharyngeal muscular dystrophy (OPMD) is characterized by slowly progressive ptosis ("oculo-"), or droopy eyelids, and dysphagia ("-pharyngeal"), or difficulty swallowing, and typically affects individuals over the age of 40. As was seen in this patient, OPMD can present complications of dysphagia like aspiration pneumonia and malnutrition (e.g. weight loss) due to swallowing difficulties. As the disease progresses, patient may develop weakness of various facial muscles (which makes chewing, speaking, and swallowing more difficult) as well as lower limb girdle weakness, which can lead to difficulty walking. A wet voice due to salivary pooling (from dysfunctional swallowing) is also commonly noted in patients with OPMD. Some individuals with OPMD have altered executive function, though most have normal cognition. Lifespan may be shorter due to complications from swallowing difficulties (especially aspiration pneumonia). Treatment focuses on symptom management through dietary modifications (e.g. a dysphagia soft diet) and surgical interventions to address ptosis and dysphagia (e.g. eyelid surgery, gastrostomy tube).
💡For OPMD, think of an Older Person (>40 years) with Mastication difficulties and Droopy eyelids.
Molecular mechanism
OPMD results from expansions of a GCN repeat (which encodes a polyalanine tract) within the first exon of the gene PABPN1. PABPN1 encodes the Poly(A)-binding nuclear protein 1, which helps add poly(A) tails to mRNA. Normally, people have 10 alanine repeats at this location, whereas people with OPMD have between 11-18 alanine repeats. In general, patients with larger polyalanine repeat sizes tend to have earlier and more severe symptoms, as is the case for many trinucleotide repeat disorders.
Diagnosis and Inheritance
The patient in Question 67 had a non-diagnostic whole exome sequencing (WES), which would be the expected result in a patient with OPMD. For technical reasons, most WES platforms are not validated to quantify trinucleotide repeat length. Instead, the diagnosis of OPMD is made with trinucleotide repeat length testing specific for PABPN1, which must be ordered as a separate test. OPMD is inherited in an autosomal dominant manner (Question 68), and most patients with OPMD have an affected parent. Of note, OPMD is one of the few trinucleotide repeat disorders that does not exhibit genetic anticipation.
💡Do you have an adult patient with Ptosis And BronchoPNeumonia (from aspiration)? If so, think about testing for a repeat expansion in PABPN1.
Incorrect answers
Blepharophimosis syndrome, also known as Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES), is characterized by the triad of blepharophimosis (narrowing of the eye opening), ptosis, and epicanthus inversus (a skin fold arising from the lower eyelid and ascending to the upper eyelid that covers the inner corner of the eye). BPES is inherited in an autosomal dominant manner and is caused by heterozygous variants in the FOXL2 gene. In contrast to the patient in this case, BPES-related symptoms are present from birth and are non-progressive. The presence of dysphagia and aspiration would also not be consistent with BPES.
Kearns-Sayre Syndrome (KSS) is a multi-systemic mitochondrial disorder characterized by progressive external ophthalmoplegia (manifesting as difficulty moving the eyes), pigmentary retinopathy (manifesting as vision loss), heart block, and muscle weakness, among other symptoms. While KSS can involve ptosis and muscle weakness, symptoms usually appear before age 20. Exome sequencing that includes mitochondrial DNA sequencing (as was performed in this patient) would most commonly show a single 4.9 kb-deletion in the mitochondrial DNA.
Optic atrophy type 1 (OPA1) is characterized by optic nerve atrophy, leading to progressive and irreversible vision loss in children and young adults. OPA1 is caused by variants in the nuclear-encoded OPA1 gene, whose protein product is involved in mitochondrial membrane fusion. The absence of visual field defects and the older age of onset of symptoms in this patient points away from this diagnosis.
Leber Hereditary Optic Neuropathy (LHON) is also a mitochondrial disorder characterized by sudden, painless vision loss and that usually affecting young adults. Patients with LHON most commonly have a point mutation (m.11778G>A) within the mitochondrial genome, which encodes a protein involved in complex I of the mitochondrial respiratory chain. As was the case for OPA1, the absence of visual field defects strongly points away from the diagnosis of LHON. LHON also does not typically manifest with ptosis or significant dysphagia, as was the case in this patient's presentation.
Learning objective
Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant disorder that presents with ptosis, dysphagia, and weight loss in middle-aged and elderly adults. OPMD is due to expansion of a GCN trinucleotide repeat in the first exon of the gene PABPN1. In contrast to other ophthalmic disorders that affect the optic nerve and/or retina, OPMD does not typically affect visual acuity. Whole exome sequencing (WES) is not able to diagnose OPMD. Rather, trinucleotide repeat length testing for PABPN1 is necessary for a definitive diagnosis.
2023 ABMGG General Exam Blueprint | V. Single Gene Inheritance → d. Single Gene Disorders → v) Neurogenetic disorders & vii) Ophthalmic genetic disorders
2023 ABGC Exam Content Outline | Domain 1. Clinical Information, Human Development, and Genetic Conditions → C. Genetic Conditions → 1. Clinical features, 6. Diagnostic processes including clinical criteria and testing strategy, 7. Mode of inheritance, & 8. Etiology
References
Oculopharyngeal Muscular Dystrophy (GeneReviews)
OPMD (Muscular dystrophy association)