


2023.11.25 | Questions 69-70
Ophthalmic genetics 👁️ (3/3) - A child with nystagmus

Hello,
I hope that you had a nice Thanksgiving! 🦃 This post is the 3rd in a series of 3 posts related to ophthalmic genetics and focuses on a child with nystagmus. I also uploaded a YouTube video related to the topic of this post.
As was mentioned in my previous post, Lynsey Rodriguez, MS, LCGC and I are currently running a board review course for those taking the ABGC board exam next month or in March 2024 (yes, the ABGC exam timelines have changed for this exam cycle!). Lynsey and I updated the course schedule (see this LinkedIn post for details) to accommodate the new exam schedule. Please reach out to us if there are any questions about the course.
If you would like to support my work, consider sharing this newsletter with folks in your network who might be interested in learning more about genetics and rare disease.
Please feel free to reach out with any feedback or ideas for future posts (daniel@studyrare.com).
-Daniel
Questions
Question 69
A 7-day-old boy is born at term. His newborn screening results come back and show elevated galactose, normal galactose-1-phosphate uridylyltransferase (GALT) activity, & decreased galactokinase activity. He appears well & is started on the appropriate therapy. In addition to seeing a metabolic physician, he should be referred to the ophthalmologist to monitor for which of the following complications?
Question 70
The patient in Question 69 is lost to follow up and presents 3 years later. Developmentally, he speaks in 3-word sentences, has a 1000-word vocabulary, and does not make eye contact. He walks independently though he often trips and walks into obstacles. His growth parameters are within normal limits. Physical exam shows nystagmus, and a slit lamp exam shows bilateral cataracts. The remainder of his exam is unremarkable. Which of the following interventions could have prevented the formation of his cataracts?
Explanation
Q69: Cataracts
Q70: Soy-based formula
This patient has galactokinase deficiency, a metabolic disorder characterized by the development of bilateral cataracts (when untreated) in an infant or child who otherwise appears well with typical growth and development. Cataracts impair vision, and can manifest in a child as nystagmus and/or frequent tripping (due to visual impairment), as seen in the patient in Question 70. Patients with galactokinase deficiency can be identified on newborn screening (see ACMG algorithm here), as was the case with this patient. If galactokinase deficiency is confirmed (by enzyme activity assay or molecular sequencing), patients should be referred to an ophthalmologist to monitor for the development of cataracts (Question 69). This is important because if left untreated, infantile-onset cataracts can lead to delayed visual development and nystagmus, as seen in this patient. Restricting dietary galactose by switching to a soy-based formula (Question 70), which does not contain galactose, helps prevent (or in some cases even reverse) the formation of cataracts. If necessary, cataract surgery can also be performed (even in early infancy) to help preserve vision. Outside of the risk for cataract formation, patients with galactokinase deficiency are typically healthy without cognitive or growth impairments. Note that galactokinase deficiency is one of several types of galactosemias (see table below), which are disorders characterized by elevated levels of galactose in the blood.

💡Patients with any type of galactosemia are at risk for cataracts (“galasses” 👓 for galactosemia)
💡Galactokinase deficiency (along with the other galactosemias) is one of the few situations where breastfeeding is contraindicated!
What is galactosemia?
The word “galactosemia” is a descriptive term that means “elevated galactose in the blood (-emia).” Galactosemia is caused by a deficiency of any one of four main enzymes involved in galactose metabolism: galactose mutarotase (GALM), galactokinase (GALK), galactose-1-phosphate uridyl transferase (GALT), and galactose epimerase (GALE). The goal of galactose metabolism is to generate the molecule glucose-1-phosphate, which enters glycolysis and generates electron carriers to help make ATP. Galactose is a dietary sugar similar to glucose that is mainly obtained as lactose, a disaccharide found in human breast milk and dairy products. In patients with galactosemia, excess galactose is converted to galactitol, a substance that accumulates in the lens of the eye, resulting in cataracts. Cataracts are thus a common feature of all of the 4 subtypes of galactosemias (see table above).
Galactose metabolism occurs via 4 enzymatic steps (see figure below)
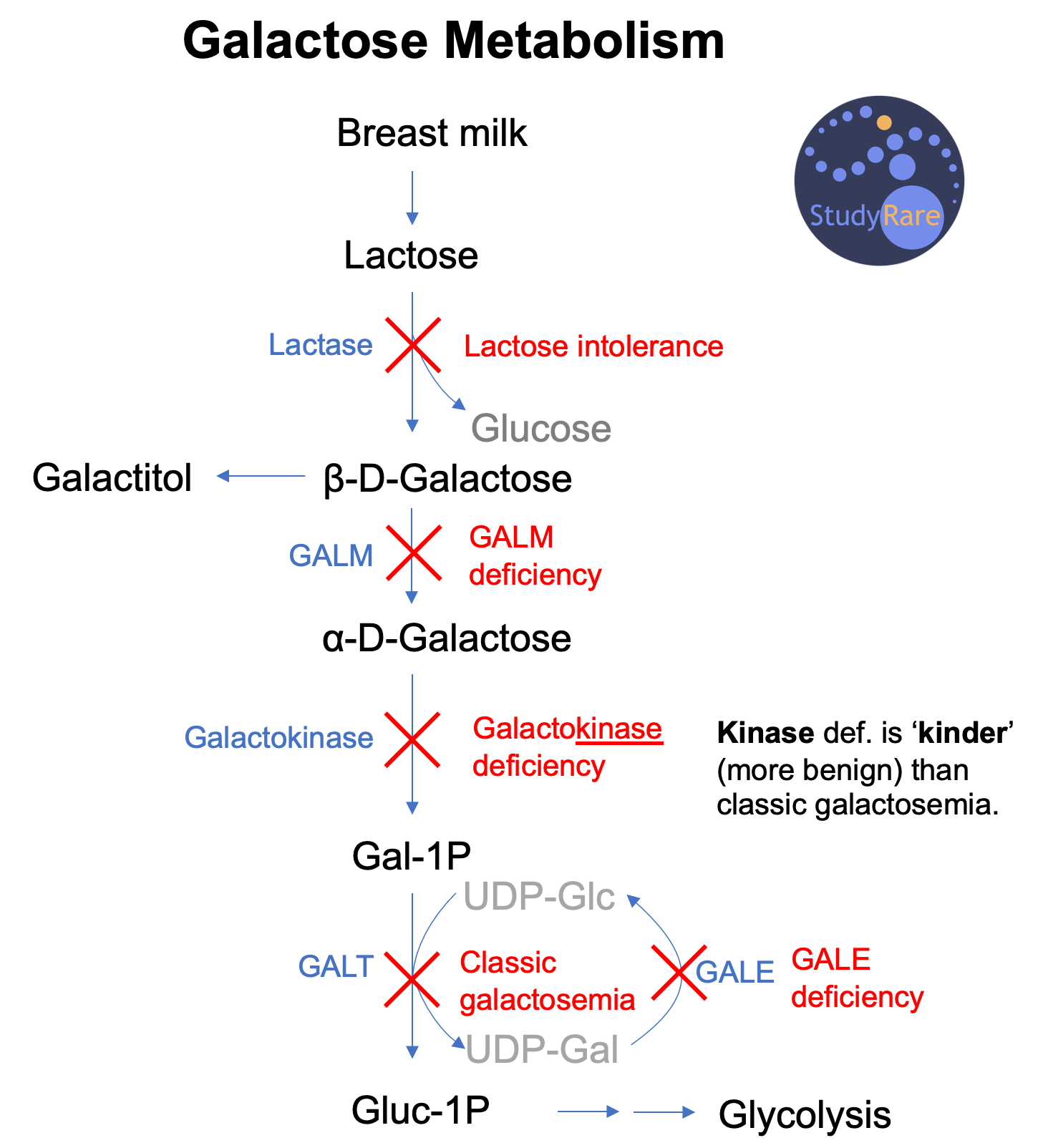
Here is a way to remember the 4 enzyme deficiencies that result in galactosemia

Other causes of cataracts
Cataracts can be divided into isolated and syndromic causes of cataracts. In addition to galactokinase deficiency, causes of isolated cataracts include pathogenic variants in the crystallin genes (which encode for the crystallin proteins that are abundant in the lens of the eye). Isolated cataracts are also common as people age (>50% by age 80 years), and most cases are multifactorial and without an identifiable genetic etiology. Syndromic causes of cataracts include Down syndrome, neurofibromatosis type 2, and congenital infections such as rubella, toxoplasmosis, and cytomegalovirus (‘sight’-omegalovirus).
💡 ‘Crystallens’ (crystallins) are abundantly expressed in the lens of the eye.
Incorrect answers
(Question 69) Corneal clouding can be seen in Hurler syndrome. Retinitis pigmentosa is a feature of Leber Congenital Amaurosis (LCA), which was discussed in a previous post. Lens dislocation is characteristic of Marfan syndrome and homocystinuria.
(Question 70) Enzyme replacement therapy is not currently available for galactosemia, though there are currently some pre-clinical trials involving gene replacement therapy for galactosemia. Supplementation of riboflavin, a B-vitamin, helps prevent vision loss and optic nerve atrophy in patients with riboflavin transporter deficiency. Corrective lenses (i.e. “glasses”) would not prevent the formation of cataracts though can help improve visual acuity in patients with or without galactosemia.
Learning objective
Galactokinase deficiency is a type of galactosemia that classically presents with cataracts in infancy without other systemic involvement. The diagnosis of galactosemia is often made on newborn screening, and a definitive diagnosis requires an enzyme activity assay and/or molecular sequencing. Management of galactokinase deficiency is with restriction of dietary galactose to prevent (or reverse) the formation of cataracts and with regular ophthalmologic screening. The differential diagnosis of congenital cataracts includes congenital infections and pathogenic variants in the crystallin genes.
P.S. For questions on classic galactosemia, see questions 7-8 from this newsletter.
2023 ABMGG General Exam Blueprint | V. Single Gene Inheritance → d. Single Gene Disorders → vii) Ophthalmic genetic disorders
2023 ABGC Exam Content Outline | Domain 1. Clinical Information, Human Development, and Genetic Conditions → C. Genetic Conditions → 1. Clinical features, 6. Diagnostic processes including clinical criteria and testing strategy, 7. Mode of inheritance, & 8. Etiology