
2026.03.01 | Questions 102-103
Gastrointestinal (3/3): Wilson disease (+ 2026 ABMGG Bootcamp Registration!)

Hello,
This is the 3rd post in our series on gastrointestinal genetics. This post focuses on Wilson disease, an important cause of liver disease that can present in childhood or adolescence and is one of the few hepatic conditions that is treatable if caught early.
We are also excited to announce that registration is now open for our ABMGG board review bootcamp, which will take place between March 17th - June 4th 2026. This bootcamp is for clinical and laboratory geneticists taking the ABMGG general exam in August 2026. We will cover topics from all 11 domains and 115 subdomains on the ABMGG content outline. Over 30 hours of live instruction plus access to a QBank (available starting May 1st) that simulates the exam environment will be provided. An early-bird discount is available through March 16th. For more information and to register, please visit our website.

If you would like to support our work, the best way to do so is to share this post with others who are preparing for genetics board exams.
Please feel free to reach out (daniel@studyrare.com) with any questions about the post, suggestions for future topics, or our upcoming courses. I hope you have a great week!
-Daniel
Questions
Question 102
A previously healthy 15-year-old boy is evaluated for progressive tremor, dysarthria, and declining school performance over the past 8 months. Physical examination shows hepatomegaly. Laboratory studies show elevated AST and ALT and low ceruloplasmin. Which of the following findings on liver biopsy would be most consistent with the suspected diagnosis?
Question 103
Genetic testing is performed for the patient in Question 102 and shows biallelic pathogenic variants in ATP7B. His 11-year-old asymptomatic sister undergoes genetic testing and is found to carry the same two variants. Which of the following is the most appropriate management for the sister?
Explanation
Question 102: Copper accumulation in hepatocytes
Question 103: Zinc therapy
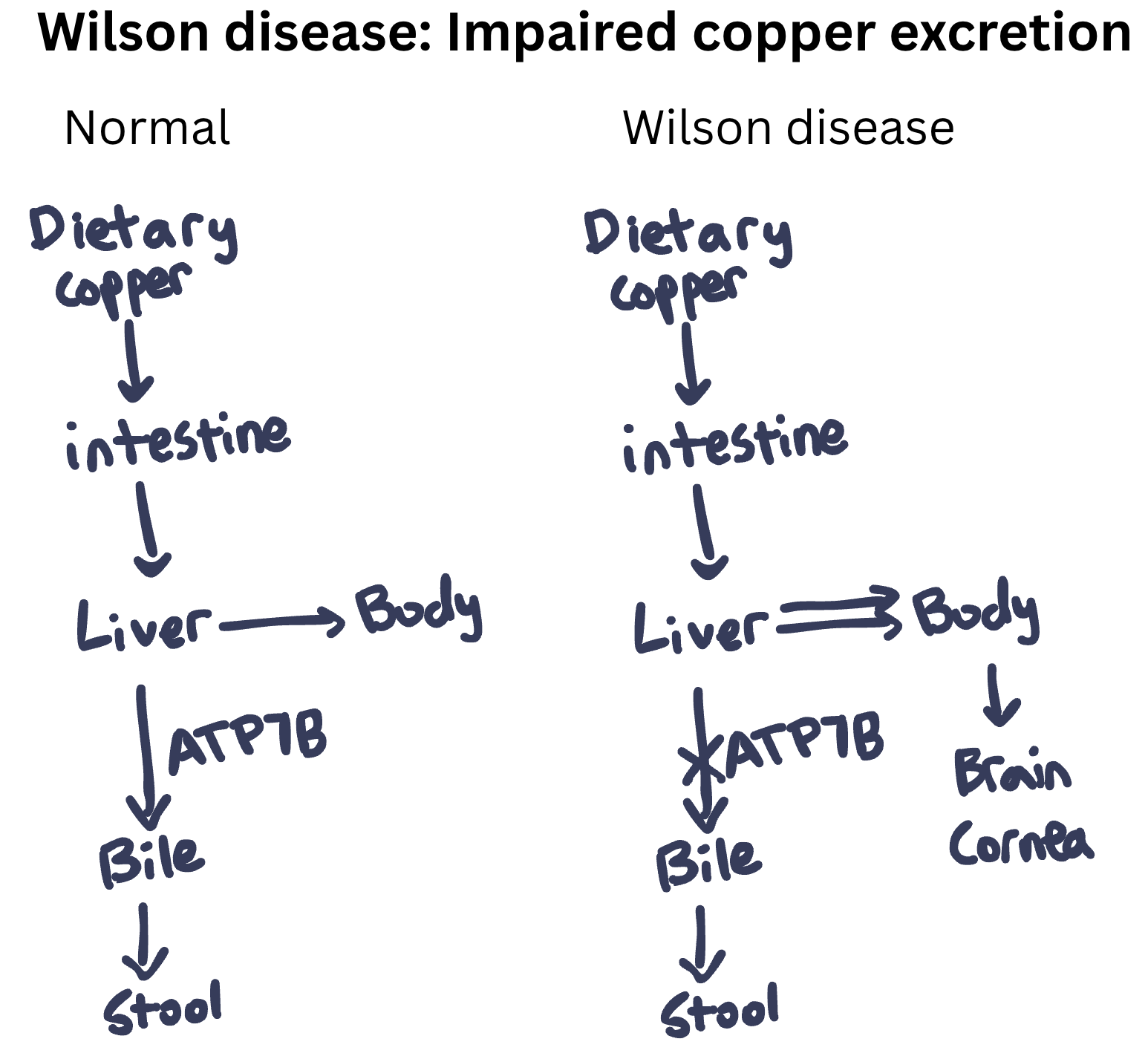
The patient in Question 102 has Wilson disease, a disorder due to excess copper in the body. Wilson disease is caused by biallelic pathogenic variants in the ATP7B gene, which encodes a copper-transporting ATPase primarily expressed in the liver. This transporter has two main functions:
Excretion of copper into bile for elimination from the body
Incorporation of copper into ceruloplasmin (the main copper-carrying protein in blood)
When ATP7B is defective, copper cannot be excreted into bile and instead accumulates in hepatocytes (Question 102). This causes hepatomegaly and cirrhosis. Over time, copper spills into the bloodstream and deposits in other organs, particularly the brain (e.g., basal ganglia), cornea, and kidneys. Symptoms are progressive if the disease is untreated. This progressive deposition of copper in both liver and brain gave rise to the other name for this disease, “hepatolenticular degeneration” (hepato = liver, lenticular = lentiform nucleus, part of the basal ganglia).
Clinical Presentation
Wilson disease typically presents between ages 5 and 35 years. There are two main presentations, and the presentation depends on which organ is primarily affected:
Hepatic presentation (more common in children):
Asymptomatic elevation of transaminases (AST, ALT)
Acute hepatitis
Chronic hepatitis progressing to cirrhosis
Acute liver failure (can be the first presentation)
Neuropsychiatric presentation (more common in adolescents/young adults):
Tremor (resting, intentional, or postural)
Dysarthria and drooling
Dystonia and rigidity
Parkinsonian features
Gait abnormalities
Depression and anxiety
Personality changes
Declining academic/work performance
Other manifestations:
Kayser-Fleischer rings (copper deposits in the cornea)
Renal tubular dysfunction (Fanconi syndrome)
Hemolytic anemia (Coombs-negative)
Cardiomyopathy
Arthropathy
Kayser-Fleischer (KF) rings are present in ~95% of patients with neurologic Wilson disease but only ~50% of those with the hepatic presentation. A slit-lamp examination by an ophthalmologist is required for detection, as the KF rings are often not visible to the naked eye.
Diagnosis
The diagnosis of Wilson disease is based on a combination of clinical findings and laboratory tests. Characteristic lab findings include low serum ceruloplasmin (this is because ATP7B helps add copper to ceruloplasmin, which would otherwise be degraded) and elevated 24-hour urine copper. Total serum copper is typically low because most of the copper in serum is bound to ceruloplasmin (which is low). However, the copper within tissues is high. Specifically, elevated hepatic copper content on liver biopsy is a highly specific test for Wilson disease. Genetic testing showing biallelic variants in ATP7B is confirmatory. The Leipzig scoring system combines clinical and laboratory findings to help establish the diagnosis, with a score ≥4 indicating a high likelihood of Wilson disease.
Management of Wilson Disease
The goals of treatment are to (1) remove excess copper from the body and (2) prevent re-accumulation of copper. Treatment is lifelong, as discontinuation leads to rapid copper re-accumulation and can precipitate acute liver failure.
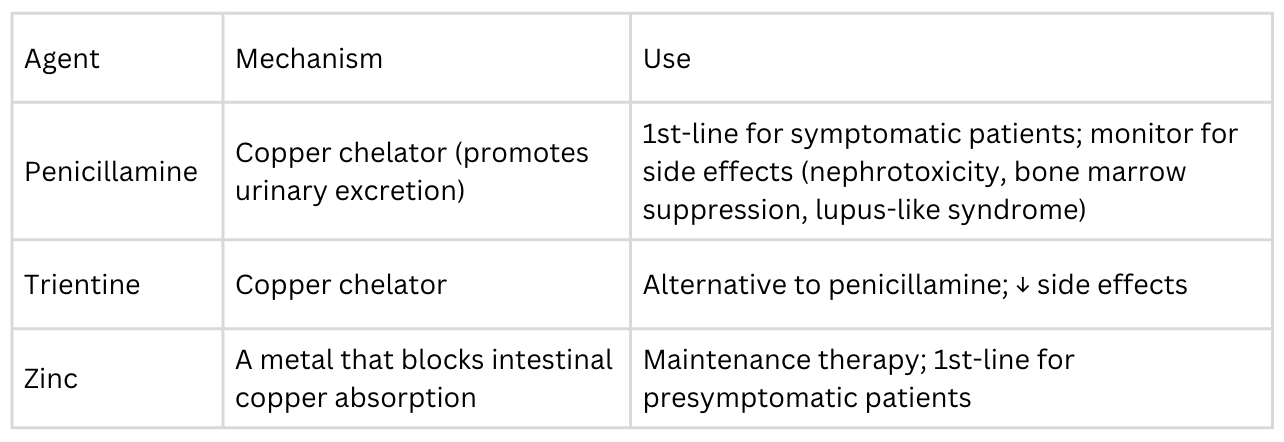
Symptomatic patients should start with chelation therapy (penicillamine or trientine) to rapidly reduce copper load, then transition to zinc for maintenance therapy. Presymptomatic patients (like the sister in Question 103) should start with zinc therapy. Zinc helps prevent absorption of copper in the first place, and presymptomatic patients should remain asymptomatic as long as they continue with this therapy. Some patients with acute liver failure may require liver transplantation (which is curative for the hepatic form, as the donor liver has functional ATP7B). Patients should also avoid high-copper foods (shellfish, liver, chocolate, nuts, mushrooms), though dietary restriction alone is not sufficient.
Incorrect Answers
Question 102
Glycogen accumulation in hepatocytes (Choice A) is characteristic of the hepatic glycogen storage diseases (GSDs) such as GSD type I (von Gierke disease). GSD I present with hepatomegaly and hypoglycemia but does not cause neuropsychiatric symptoms or low ceruloplasmin. Iron deposition in hepatocytes (Choice B) describes hereditary hemochromatosis (HFE-related iron overload). Hemochromatosis typically presents in adults, is characterized by the accumulation of iron (not copper) in the liver, and does not cause the neuropsychiatric features seen in this patient. Reduction in the number of bile ducts (Choice D) is the classic histologic finding in Alagille syndrome, which presents in infancy with cholestasis and is associated with cardiac defects, butterfly vertebrae, and characteristic facies. The patient in this question was previously healthy without known congenital anomalies.
Question 103
Dietary copper supplementation (Choice A) is incorrect. Wilson disease is caused by copper excess, not deficiency. Copper supplementation would worsen the condition. Observation only (Choice C) is also incorrect. Presymptomatic treatment prevents organ damage (liver cirrhosis, brain injury) and is strongly recommended for all individuals with biallelic pathogenic variants, such as the sister mentioned in this question. Liver transplantation (Choice D) is reserved for acute liver failure or decompensated cirrhosis unresponsive to medical therapy. This intervention is not appropriate for an asymptomatic patient.
Learning objective
Wilson disease is an autosomal recessive disorder caused by biallelic pathogenic variants in ATP7B that results in excess accumulation of copper throughout the body. It typically presents in childhood or adolescence with liver disease, neuropsychiatric symptoms (tremor, dysarthria, behavioral changes), and Kayser-Fleischer rings. Diagnosis relies on low ceruloplasmin, elevated 24-hour urine copper, and elevated hepatic copper content on biopsy. Symptomatic patients are treated with copper chelation (helps remove copper from the body), while presymptomatic individuals receive zinc therapy (helps prevent copper absorption into the body). Early treatment prevents organ damage and is associated with a favorable prognosis.
2025 ABMGG General Exam Blueprint | V. Single gene inheritance → d) Single gene disorders → x) Other
2023 ABGC Exam Content Outline | Domain 1C. Genetic Conditions. (Wilson disease is one of the conditions listed on the ABGC self-study guide)
Additional resources
GeneReviews (Wilson disease)
Wilson disease (Nature Reviews Disease Primers)
Consider reviewing our previous post on Menkes disease (Questions 60-62), the other major disorder of copper metabolism.