


2024.10.14 | Questions 86-87
Imprinting disorders (2/4) - Angelman syndrome (+ Board Review Bootcamp!)


Hello,
This is the second in a series of 4 posts related to imprinting disorders. This series of posts has been written together with Sofia Dallarda, a genetic counseling student who worked with us over the summer.
I am also excited to announce our winter bootcamp for anyone preparing for the Feb 2025 ABGC board exam! The course will run from Dec 2, 2024 - Jan 23, 2025. Early-bird registration at a discounted rate is available through November 22nd. Register here.

Please feel free to let me know if you have any comments or suggestions related to this post. I hope you have a great week!
-Daniel
Questions
Question 86
A 12-month-old girl presents to the clinic for an evaluation. She has global developmental delay and difficulty feeding. She is at the 5th percentile for weight and height and at the 2nd percentile for head circumference. She meets the clinical diagnostic criteria for Angelman syndrome. DNA methylation analysis at 15q11-q13 is sent and is non-diagnostic. Which of the following is the next best step in diagnosis?
Question 87
Genetic testing is sent for the patient in Question 86 and is positive for a pathogenic loss-of-function variant in UBE3A. Which of the following pieces of anticipatory guidance is most appropriate to share with the family in this scenario?
Explanation
Question 86: UBE3A gene sequencing
Question 87: "She is at risk for seizures."
Angelman syndrome is an imprinting disorder characterized by multiple neurologic features that include developmental delays, minimal speech, gait ataxia, acquired microcephaly, and seizures (Question 87). Angelman usually presents as gross motor or speech delays in the first 6-12 months of life, and there are typically no signs of Angelman at birth. Seizures affect many patients with Angelman syndrome and typically start within the first few years of life. Behaviorally, children with Angelman may laugh and smile frequently and may seem easily excitable. Skin hypopigmentation can be seen with patients who have a deletion at 15q11.2-q13 that includes the melanin-related protein OCA2 (discussed further below). Children with Angelman syndrome most often have a structurally normal brain and will usually reach a typical adult height.
💡 Think of Angelman as a disease of the brain 🧠. Most of the major symptoms of Angelman (e.g. seizures, intellectual disability, ataxia) can be linked back to the brain in some way.
Molecular basis of Angelman syndrome
Angelman syndrome results from the loss of ubiquitin protein ligase E3A (UBE3A) expression, which plays a crucial role in the formation of neuronal synapses. In the brain 🧠, UBE3A is expressed only from the maternally-inherited allele (and not from the paternally-inherited allele). Angelman syndrome results when the maternally-inherited copy of UBE3A is altered or missing, which leaves the brain with no functional copies of UBE3A.
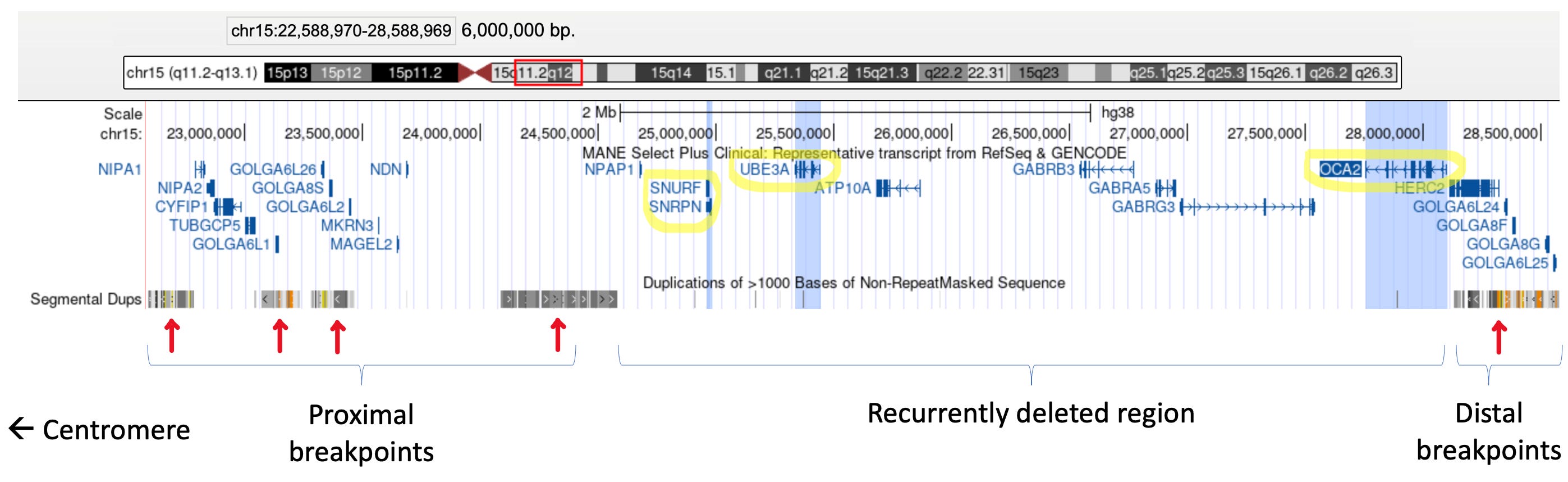
Molecular alterations of UBE3A most commonly result from a de novo 5-6 megabase deletion at 15q11.2-q13. While deletion of this region on the maternally-inherited chromosome results in Angelman syndrome, deletion of this same imprinted region on the paternally-inherited chromosome causes Prader-Willi syndrome (see our previous post). Many deletions at 15q11.2-q13 include OCA2, a non-imprinted gene involved in melanin production. Loss of OCA2 results in hypopigmentation, which is observed in patients with both Angelman and Prader-Willi syndromes when the deletion involves this gene.
Recurrent deletions and duplications at 15q11.2-q13
Recurrent deletions and duplications with similar breakpoints occur at the 15q11.2-q13 region in unrelated individuals. But why does this happen? This is because this region is flanked by segmental duplications (aka low copy repeats, or LCRs), which are highly similar repetitive sequences (see image below). The presence of these LCRs can result in non-allelic homologous recombination (NAHR), which happens when the LCRs misalign in meiosis and cause unequal crossing over between homologous chromosomes. This can cause a deletion and duplication of the genetic material between the LCRs (see this paper for an illustration). NAHR at 15q11.2-q13 is thus the mechanism behind the deletions seen in Prader-Willi and Angelman and the interstitial duplications seen in Dup15q syndrome.
Diagnosing Angelman syndrome
The definitive diagnosis of Angelman is based on molecular testing. The three most common molecular changes that lead to Angelman include:
Deletions of 15q11.2-q13 (~70% of cases)
Paternal uniparental disomy (UPD) of chromosome 15 (~5% of cases)
UBE3A pathogenic variants (~10% of cases)
Molecular testing identifies a genetic etiology in about 90% of people with Angelman syndrome. The first step in molecular testing for Angelman syndrome is DNA methylation analysis, which would flag positive in the presence of a deletion or paternal UPD. If the methylation analysis returns normal, UBE3A gene sequencing should be sent next (Question 86). Of note, pathogenic UBE3A variants that are inherited from an unaffected mother can result in children with Angelman syndrome (50% recurrence risk). A diagnostic flowchart is available through the Angelman syndrome foundation, and an illustration of the 3 molecular mechanisms is shown below.

Genotype-phenotype relationship
In Angelman syndrome, patients with large megabase-sized deletions at 15q11.2-q13 are often the most severely affected, while patients with paternal uniparental disomy (UPD) have fewer and more mild symptoms (e.g. no microcephaly, fewer seizures). Patients with imprinting defects and UBE3A variants may fall somewhere in the middle of this spectrum. Note that there is a high degree of clinical variability even among patients with the same underlying disease mechanism.

Management
Management of Angelman syndrome is supportive and requires a multidisciplinary team of clinicians including neurology (for seizures, motor delays, and ataxia), physical therapy, speech therapy, nutrition, and ophthalmology (if strabismus is present), among other specialists. A table listing management guidelines by age is available in this paper (Table 1). Clinical trials involving antisense oligonucleotide therapy that activates the paternal copy of UBE3A are in progress (see full list here), though no targeted therapeutics are currently available.
Incorrect answers
Question 86
Microsatellite studies (choice A) are used to assess parental origin in the setting of uniparental disomy (UPD). Once the DNA methylation analysis is negative (as was the case for this patient), UPD is virtually excluded as a cause of Angelman syndrome. Therefore, microsatellite studies would not provide further diagnostic clarity in this case. Fluorescence in situ hybridization (FISH) (choice C) is used to detect chromosomal deletions, duplications, or rearrangements. Since the DNA methylation analysis (which flags positive with most chromosomal deletions) was negative, FISH is unlikely to uncover additional findings and is not the most appropriate next step. While DNA methylation testing of the mother (choice D) would be normal and would not provide additional information in this scenario, testing her for the presence of a UBE3A variant could be considered once a variant is identified in the proband. Alterations such as DNA methylation that do not permanently change the DNA sequence are not typically heritable.
Question 87
Cardiac arrhythmias (choice A) are not characteristic of Angelman syndrome (AS) though are a feature of long QT syndrome and Brugada syndrome, among others. Progressive hearing loss (choice B) and progressive vision loss (choice D) are not features of AS though can be seen in Usher syndrome and mitochondrial disorders such as progressive external ophthalmoplegia (PEO). While speech delays can be secondary to hearing loss, the speech delay in AS is not due to hearing loss but rather due to differences in how the brain functions. Some patients with AS may have strabismus or refractive errors that affect vision, though this would not result in progressive vision loss.
Learning objective
Angelman syndrome is an imprinting disorder characterized primarily by neurologic symptoms including seizures, intellectual disability, ataxia, and gross motor delays. On a molecular level, loss of UBE3A expression from the maternally-inherited allele results in Angelman syndrome. UBE3A plays an important role in neurons and synapse formation, which explains the primarily neurologic features of this disorder. The diagnosis of Angelman is typically made with DNA methylation testing or UBE3A sequencing. Management is supportive, and clinical trials involving targeted therapeutics to increase UBE3A expression are ongoing.
2023 ABGC Exam Content Outline | Domain 1. Clinical Information, Human Development, and Genetic Conditions → C. Genetic Conditions → 8. Etiology | Domain 3. Testing Interpretation, Testing Options, and Reproductive Risk Management → B. Testing Options → 1. Diagnostic
2025 ABMGG General Exam Blueprint | V. Single gene inheritance → c. Atypical inheritance → ii) Parent of origin effects on inheritance (genomic imprinting)
References
GeneReviews (Angelman Syndrome)
Educational resources from the Angelman Syndrome Foundation
 | A guest post by
|